- Pharmacocinetique
-
Pharmacocinétique
La pharmacocinétique, qui suit la phase biopharmaceutique, a pour but d'étudier le devenir d'une substance active contenue dans un médicament dans l'organisme. Elle comprend quatre phases, se déroulant simultanément :
- absorption ;
- distribution ;
- métabolisme ;
- élimination de la substance active.
La pharmacocinétique peut aussi concerner le devenir de substances chimiques quelconques dans le corps. Lorsque ces substances sont toxiques, on utilise alors le terme de 'toxicocinétique'.
La détermination des paramètres pharmacocinétiques d'une substance active apporte les informations qui permettent de choisir les voies d'administration et la forme galénique, et d'adapter les posologies pour son utilisation future.
Sommaire
Pharmacocinétique descriptive : les processus ADME
Le sigle ADME renvoie à quatre processus : absorption, distribution, métabolisme et élimination.
Absorption
La substance active doit traverser les membranes biologiques du site d'absorption vers le sang pour pénétrer dans la circulation systémique.
Quatre points sont à considérer en particulier :
- Les obstacles à franchir pour passer dans la circulation systémique : il s'agit des membranes biologiques, de type lipidique, comme :
- la paroi du tube digestif lors de l'administration de comprimés ;
- la peau lors de l'administration de pommades ;
- la paroi du rectum lors de l'administration de suppositoires ;
- etc.
- Les conditions d'absorption relatives au médicament absorbé :
- la substance active doit être suffisamment hydrosoluble pour être dissoute ;
- la substance active doit être suffisamment liposoluble pour franchir les barrières biologiques lipophiles ;
- la substance active doit être sous forme neutre pour franchir les barrières biologiques ;
- la taille des molécules du principe actif peut aussi avoir son importance ;
- etc.
- Conditions d'absorption relatives au milieu :
- absorption dans l'estomac : les substances actives acides faibles sont absorbées au niveau de l'estomac, car elles ne sont pas ionisées en milieu acide ; sous forme non-ionisée liposoluble, elles sont par conséquent capables de franchir les barrières biologiques lipophiles. Les substances actives bases faibles sont elles faiblement absorbées au niveau de l'estomac, car elles sont ionisées en milieu acide.
- absorption au niveau de l'intestin grêle (milieu plutôt basique). Les substances actives acides faibles sont faiblement absorbées au niveau de l'intestin grêle, car elles sont ionisées en milieu basique ; sous forme ionisée hydrosoluble, elles franchissent difficilement les barrières biologiques lipophiles. Les substances actives bases faibles, elles, sont bien absorbées à ce niveau !
- l'importance de la vidange gastrique. Si elle est trop rapide, les médicaments absorbés par l'estomac n'auront pas suffisamment de temps pour franchir les parois de l'estomac ; si elle est trop lente, les médicaments absorbés par l'intestin grêle parviendront dans le milieu sanguin avec un certain retard.
- La notion de premier passage :
Lors de la prise orale d'un médicament, suivie d'une absorption gastro-intestinale, la substance active est d'abord transportée par la veine porte vers le foie avant d'atteindre la circulation systémique. La substance active peut ainsi subir une métabolisation hépatique présystémique avant d'atteindre la circulation générale : c'est l'effet de premier passage hépatique. C'est en général le plus important quantitativement, mais il existe également un premier passage pulmonaire et un premier passage digestif. L'importance de chacun est différente selon les caractéristiques physico-chimiques et biologiques de la substance en question.
L'organisation du système vasculaire permet d'éviter en grande partie l'effet de premier passage hépatique en cas d'administration par suppositoire : un tiers seulement du sang circulant dans les veines rectales passe par le foie avant d'atteindre la circulation générale. Ce premier passage est totalement évité par administration sub-linguale (médicament à faire fondre sous la langue) : la substance active est absorbée et déversée dans les veines jugulaires, aboutissant directement dans la circulation au niveau de l'arc aortique. Cette caractéristique fait une grande partie de l'intérêt de ces modes d'administration en cas d'urgence relative (un quart d'heure à une demi-heure de délai d'action).
Enfin, l'administration d'un principe actif par injection permet un passage direct dans la circulation, sans avoir à se soucier des problèmes liés à l'absorption évoqués ci-dessus. Ceci en fait la voie d'administration privilégiée dans les cas d'urgence.
Distribution
La substance active parvenue à la circulation systémique peut se lier plus ou moins fortement et de façon réversible aux protéines plasmatiques (albumine, globulines ou lipoprotéines) :
- Exemples de médicaments fortement liés aux protéines plasmatiques : dicoumarol (anticoagulant), phénytoïne (antiépiléptique) ;
- Exemples de médicaments moyennement liés aux protéines plasmatiques : phénobarbital (antiépileptique, hypnotique), théophylline (antiasthmatique)... ;
- Exemples de médicaments faiblement liés aux protéines plasmatiques : digoxine (cardiotonique), paracétamol (analgésique, antipyrétique)...
En fonction de ses propriétés physico-chimiques et biochimiques, la substance active peut également s'accumuler dans certains organes ou tissus contenant ou non des récepteurs pharmacologiques :
- affinité pour les protéines tissulaires ;
- affinité pour les acides nucléiques ;
- affinité pour les graisses ;
- affinité pour les tissus cibles contenant les récepteurs pharmacologiques.
On peut notamment calculer le volume de distribution de la substance pour caractériser la distribution dans le corps humain.
Métabolisme
C’est la transformation du médicament par le système enzymatique de l’organisme, en particulier au niveau du foie (organe contenant beaucoup d’enzymes).
Les métabolites ainsi produits peuvent alors avoir :
- aucune activité (directement éliminés) ;
- une activité médicamenteuse avant élimination ;
- une activité toxique avant élimination.
Les substances métabolisées peuvent être transformées en un autre produit actif. Ce phénomène peut être exploité pour concevoir un médicament inactif sous sa forme administrée au patient, mais conduisant à des métabolites actifs. On parle alors de médicament pro-drogue. Cet artifice peut être particulièrement utile quand la forme active de la substance active présente des difficultés d'administration ou d'absorption telle quelle.
Élimination
Lorsqu'elle est présente dans la circulation générale, la substance active du médicament a tendance à être évacuée par l'organisme, qui utilise de nombreux mécanismes d'élimination (excrétion et/ou métabolisation). La substance active est éliminée par l'organisme soit sous forme inchangée, soit sous forme d'un ou de plusieurs métabolites généralement inactifs, soit encore sous les deux formes, dans des proportions variables. Cette “auto-épuration” de l'organisme se quantifie par le paramètre de clairance.
La clairance (CL)
Tout organe présente un courant d'entrée (flux artériel) et un courant de sortie (flux veineux). La présence d'une concentration médicamenteuse moindre au niveau veineux qu'au niveau artériel prouve que l'organe a éliminé une partie du médicament. Cette capacité d'épuration de l'organe correspond au processus de la clairance. On définit généralement la clairance sanguine ou plasmatique d'un médicament par un organe comme le volume sanguin ou plasmatique totalement débarrassé de la substance par unité de temps. Ce paramètre correspond à un débit exprimé en ml/min
La clairance est caractérisée par :
- la concentration du médicament à l'entrée de l'organe CA (sang artériel) ;
- la concentration du médicament à la sortie de l'organe CV (sang veineux) ;
- le débit sanguin Q de l'organe concerné.
Dès lors, la réaction médicament/organe s'exprime en termes de clairance selon la relation suivante :
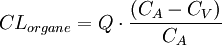
La clairance totale
L'épuration d'un médicament par l'organisme n'est généralement pas le fait d'un seul et unique organe. En effet, les possibilités pour l'organisme de se débarrasser d'une substance médicamenteuse sont multiples.
Deux mécanismes peuvent néanmoins être envisagés :
- l'élimination directe du composé ;
- l'élimination du composé après biotransformation (métabolisation).
La métabolisation d'une substance médicamenteuse est en règle générale principalement due au foie. Cependant, de nombreux autres organes peuvent y jouer un rôle : poumons, intestin ou cerveau.
Les voies d'excrétion sont elles aussi multiples. De manière logique, les voies prédominantes sont généralement la voie urinaire ou la voie biliaire (fécès). Cependant, de nombreux autres organes peuvent participer à cette élimination : les poumons, la peau (transpiration), les glandes salivaires, les glandes lacrymales...
Ainsi, un médicament subit généralement plusieurs processus d'épuration. La clairance totale correspond à la somme des clairances partielles (rénale, hépatique, intestinale, pulmonaire,...). Très schématiquement, on distingue la clairance rénale et la clairance extrarénale.
La clairance extrarénale est le résultat :
- de la clairance hépatique provenant de l'addition des valeurs de clairance biliaire et de la clairance métabolique hépatique ;
- des différentes clairances métaboliques liées à l'activité de biotransformation des poumons, des intestins ou d'autres organes.
En résumé, pour l'élimination
Le foie est le site principal de la biotransformation de la substance active : métabolisation de la substance active en dérivés hydrosolubles, actifs ou inactifs. Dans une moindre part, il constitue un organe d'excrétion (élimination biliaire).
Le rein est le principal organe d'excrétion de la substance active inchangée et des métabolites plus hydrosolubles :
- filtration glomérulaire de la fraction libre du médicaments métabolisé ou non ;
- sécrétion tubulaire des formes cationiques ;
- réabsorption.
Pharmacocinétique analytique : concepts et modèles
Influence de la dose administrée
L'effet pharmacologique ou thérapeutique d'un médicament est davantage lié à la valeur des concentrations plasmatiques qu'à la quantité totale présente dans l'organisme ou à la quantité absorbée. Partant de ce principe, on peut définir :
- un seuil thérapeutique : concentration minimale en-dessous de laquelle aucune activité n'est obtenue ;
- une limite supérieure : concentration maximale au-delà de laquelle apparaissent des effets indésirables ;
- un intervalle thérapeutique : zone intermédiaire dans laquelle les concentrations sont à la fois actives et non-toxiques.
L'importance de l'écart entre les concentrations actives et toxique est variable. Cet écart est appelé index thérapeutique. Ainsi, on distingue les médicaments à index thérapeutiques élevé et les médicaments à index thérapeutique faible. Pour ces derniers, les posologies d'administration doivent être bien définies afin que les concentration plasmatiques restent strictement dans l'intervalle thérapeutique. C'est le cas du lithium, de la phénytoïne, de la digoxine, de la théophylline...
La pharmacocinétique d'une substance médicamenteuse peut être fortement influencée par l'alimentation, par les variables physiologiques comme l'insuffisance rénale ou l'insuffisance hépatique, par l'âge du patient et également par les médicaments pris en co-médication.
Influence de la fréquence d'administration
Les médicaments font l'objet, d'un manière très générale, d'administrations réitérées. Les objectfs à atteindre sont les mêmes que lors d'une administration unique :
- obtenir une efficacité thérapeutique dans les meilleurs délais ;
- maintenir en permanence une concentration plasmatique active dans l'intervalle thérapeutique ;
- éviter tout phénomène d'accumulation pouvant conduire à l'apparition d'effets toxiques.
Partant du principe de base que la dose choisie est bien adaptée, la fréquence d'administration est le facteur fondamental permettant de respecter les exigences énoncées. Les concentrations plasmatiques vont osciller entre deux valeurs, l'une maximale et l'autre minimale. Pour une efficacité optimale, la concentration maximale doit être inférieure au seuil toxique et la concentration minimale doit être supérieure au seuil thérapeutique.
Analyse non-compartimentale
L'analyse de données de pharmacocinétique ou la prédiction de propriétés pharmacocinétiques peut se faire à l'aide de méthodes compartimentales (qui divisent conceptuellement le corps en une série de boites homogènes) ou non-compartimentales. Les méthodes non-compartimentales estiment typiquement l'exposition à une substance sur la base de l'aire sous la courbe d'un graphique de l'évolution temporelle de la concentration de substance dans le sang.
Analyse compartimentale
Les méthodes compartimentales cherchent à décrire directement l'évolution temporelle simultanée des concentrations de la substance dans différentes parties du corps (organes, tissus ou cellules.) Ces méthodes sont complémentaires des méthodes expérimentales décrites ci-dessus en ce qu'elles permettent de mieux interpréter leurs résultats et de les extrapoler à des conditions non-observées ou même non-observables (par exemple chez la femme enceinte.)
On différencie les modèles compartimentaux "classiques" et les modèles pharmacocinétiques "physiologiques" (dits "modèles PBPK".)
Articles connexes
 Portail de la médecine
Portail de la médecine Portail de la pharmacie
Portail de la pharmacie Portail de la biochimie
Portail de la biochimie
Catégories : Pharmacologie | Médecine
Wikimedia Foundation. 2010.